Quantitative Structure Activity Relationship
Introduction
Quantitative Structure Activity Relationships (QSARs) are mathematical models that are used to predict measures of toxicity from physical characteristics of the structure of chemicals (known as molecular descriptors). Acute toxicities (such as the concentration that causes half of a fish population to die) are one example of the toxicity measures that may be predicted from QSARs. Simple QSAR models calculate the toxicity of chemicals using a simple linear function of molecular descriptors:
Toxicity = ax1+bx2+c
where x1 and x2 are the independent descriptor variables and a, b, and c are fitted parameters. Examples of molecular descriptors include the molecular weight and the octanol-water partition coefficient. Additional examples are provided in our Molecular Descriptors Guide (PDF) (47 pp, 279 KB).
Uses of QSAR toxicity models
- QSAR toxicity predictions may be used to screen untested compounds in order to establish priorities for traditional bioassays, which are expensive and time-consuming.
- QSAR models are useful for estimating toxicities needed for green process design algorithms such as the Waste Reduction Algorithm.
Objectives
- Develop quantitative structure activity relationship (QSAR) methodologies to estimate toxicity from molecular structure
- Develop software, such as the Toxicity Estimation Software Tool (T.E.S.T.), that will enable users to easily estimate toxicity from molecular structure
QSAR Methodologies
Several QSAR methodologies have been developed:
- Hierarchical method - The toxicity for a given query compound is estimated using the weighted average of the predictions from several different models. The different models are obtained by using Ward’s method to divide the training set into a series of structurally similar clusters. A genetic algorithm-based technique is used to generate models for each cluster. The models are generated prior to runtime.
- FDA method - The prediction for each test chemical is made using a new model that is fit to the chemicals that are most similar to the test compound. Each model is generated at runtime.
- Single-model method - Predictions are made using a multilinear regression model that is fit to the training set (using molecular descriptors as independent variables) using a genetic algorithm-based approach. The regression model is generated prior to runtime.
- Group contribution method - Predictions are made using a multilinear regression model that is fit to the training set (using molecular fragment counts as independent variables). The regression model is generated prior to runtime.
- Nearest neighbor method - The predicted toxicity is estimated by taking an average of the three chemicals in the training set that are most similar to the test chemical.
- Consensus method - The predicted toxicity is estimated by taking an average of the predicted toxicities from each of the above QSAR methodologies.
 Random forest method- The predicted toxicity is estimated using a decision tree which bins a chemical into a certain toxicity score (i.e. positive or negative developmental toxicity) using a set of molecular descriptors as decision variables. The random forest method is currently only available for the developmental toxicity endpoint. The random forest model for the developmental toxicity endpoint was developed by researchers at Mario Negri Institute for Pharmacological Research as part of the CAESAR project.
Random forest method- The predicted toxicity is estimated using a decision tree which bins a chemical into a certain toxicity score (i.e. positive or negative developmental toxicity) using a set of molecular descriptors as decision variables. The random forest method is currently only available for the developmental toxicity endpoint. The random forest model for the developmental toxicity endpoint was developed by researchers at Mario Negri Institute for Pharmacological Research as part of the CAESAR project. 
These methodologies are explained in detail in the publications below.
Toxicity Estimation Software Tool (T.E.S.T.)
T.E.S.T. will enable users to easily estimate acute toxicity using the above QSAR methodologies. The software is now available for download. The software is described in further detail in the User's Guide (PDF) (66 pp, 540 KB).The software is based on the Chemistry Development Kit , an open-source Java library for computational chemistry.
The software includes models for the following endpoints:
- 96-hour fathead minnow 50% lethal concentration (LC50)
- 48-hour daphnia magna 50% lethal concentration (LC50)
- Tetrahymena pyriformis 50% growth inhibition concentration (IGC50)
- Oral rat 50% lethal dose (LD50)
- Bioconcentration Factor (BCF). The bioconcentration factor data set was compiled by researchers at the Mario Negri Isituto Di Ricerche Farmacologiche.
- Developmental Toxicity (DevTox).
- Ames mutagenicity (Mutagenicity).
The software now contains models for the following physical properties:
Models for additional endpoints will be added as they are completed.
 Get email alerts when new versions of the T.E.S.T. software are posted.
Get email alerts when new versions of the T.E.S.T. software are posted.
Download T.E.S.T (version 4.0):
The training and prediction sets used in the software are available here.
Sample structure data files (such as a MDL SD file) are available here.
What's new in Version 4.0?
- Physical properties are now estimated
- Batch mode is improved:
- Loading can now be interrupted
- Chemicals with loading errors are displayed at the top of the batch table
- Can now load SMILES files with no identifier field (chemicals are assigned arbitrary IDs)
- Aromaticity detection is improved:
- Can handle aromatic bond orders (bond order = 4) in mol or sd files
- The SMILES parser has been improved to better handle complicated aromatic ring systems
- Added Options screen:
- Added ability to change the output directory after it has been set
- The program now remembers the previously selected output folder
- The "Relax fragment constraint" checkbox was moved to Options screen
Prior Version History
- 3.3 (7/8/10)
- Daphnia magna LC50 endpoint was added
- AMES Mutagenicity endpoint was added
- The following changes were made for binary endpoints such as developmental toxicity and AMES mutagenicity:
- QSAR models now have stricter statistical standards (leave one out concordance = 0.8, sensitivity = 0.5, and specificity = 0.5)
- Model statistics such as concordance, sensitivity, and specificity are now displayed in the results web pages
- 3.2 (12/18/09)
- Reproductive toxicity endpoint was added
- Random forest QSAR method was added (for reproductive toxicity endpoint only)
- 3.1 (6/23/09)
- Fixed issue with running TEST in non-english speaking countries
- 3.0 (4/14/09)
- Random selection is used to divide the data sets into training and test sets
- Added BCF endpoint
- Added consensus prediction method
- 2.0 (2/24/09)
- Each toxicity data set is now split into a training and test set.
- The toxicity models included in the software are now fit to the training sets (previously they were fit to the overall sets)
- The batch mode was improved (chemicals can be added and the list can now be saved as an SDF)
- 1.0.3 (10/24/08)
- Fixed calculation of "ieadje" molecular descriptor
- Fixed definitions of chi descriptors in numbered list in molecular descriptors guide
System requirements
Installation Instructions
- Save the appropriate installation file to your hard drive. Due to the large size of the file, the download may take 15 minutes or longer depending on the speed of the connection.
- Double-click the installation file (for Linux users: open a shell, cd to the directory where you downloaded the installer and at the prompt type: sh ./install.bin).
Silent Installation Instructions for Network Administrators (for Windows users)
- The software can be installed silently by issuing the following command at the command prompt:
install -i silent
Publications
Sushko, I.; Novotarskyi1, S.; Körner, R.; Pandey, A. K.; Cherkasov, A.; Li, J.; Gramatica, P.; Hansen, K.; Schroeter, T.; Müller, K.-R.; Xi, L.; Liu, H; Yao, X.; Öberg, T.; Hormozdiari, F.; Dao, F.; Sahinalp, C.; Todeschini, R.; Polishchuk, P.; Artemenko, A.; Kuz’min, V.; Martin, T.M.; Young, D. M.; Fourches, D.; Muratov, E.; Tropsha, A.; Baskin, I.; Horvath, D.; Marcou, G.; Varnek, A; Prokopenko, V. V.; Tetko, I.V. (2010). “Applicability domains for classification problems: benchmarking of distance to models for AMES mutagenicity set.” J. Chem. Inf. Model, 50, 2094-2111.
Cassano, A.; Manganaro, A; Martin, T.; Young, D.; Piclin, N.; Pintore, M.; Bigoni, D.; Benfenati, E. (2010). “The CAESAR models for developmental toxicity.” Chemistry Central Journal, 4(Suppl 1):S4.
Zhu, H.; Martin, T.M.; Young, D. M.; Tropsha, A. (2009). “Combinatorial QSAR Modeling of Rat Acute Toxicity by Oral Exposure.“ Chemical Research in Toxicology, 22 (12), pp 1913-1921.
Benfenati, E., Benigni, R., Demarini, D.M., Helma, C., Kirkland, D., Martin, T.M., Mazzatorta, G., Ouedraogo-Arras, G., Richard, A.M., Schilter, B., Schoonen, W.G.E.J., Snyder, R.D., and C. Yang. (2009). “Predictive Models for Carcinogenicity and Mutagenicity: Frameworks, State-of-the-Art, and Perspectives.” Journal of Environmental Science and Health Part C, 27, 2: 57-90.
Young, D.M.; Martin, T.M.; Venkatapathy, R.; Harten, P. (2008) “Are the Chemical Structures in your QSAR Correct?” QSAR & Combinatorial Science, 27 (11-12), 1337-1345.
Martin,T.M., P. Harten, R. Venkatapathy, S. Das and D.M. Young. (2008). “A Hierarchical Clustering Methodology for the Estimation of Toxicity.” Toxicology Mechanisms and Methods, 18, 2: 251–266.
Martin, T.M., and D.M. Young. (2001). “Prediction of the Acute Toxicity (96-h LC50) of Organic Compounds in the Fathead Minnow (Pimephales Promelas) Using a Group Contribution Method.” Chemical Research in Toxicology, 14, 10: 1378–1385.
Contact
Todd Martin, PhD.
Research Chemical Engineer








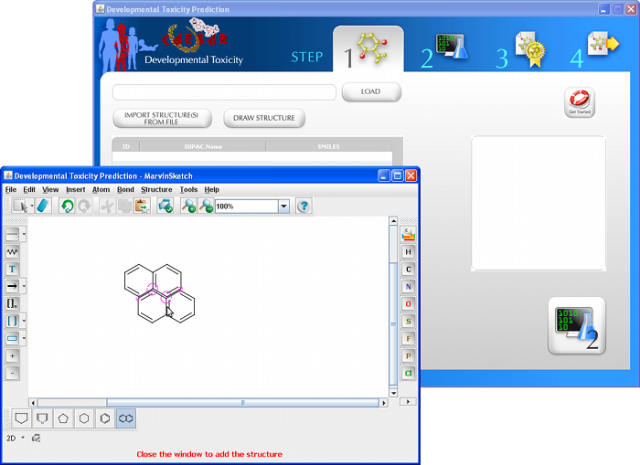

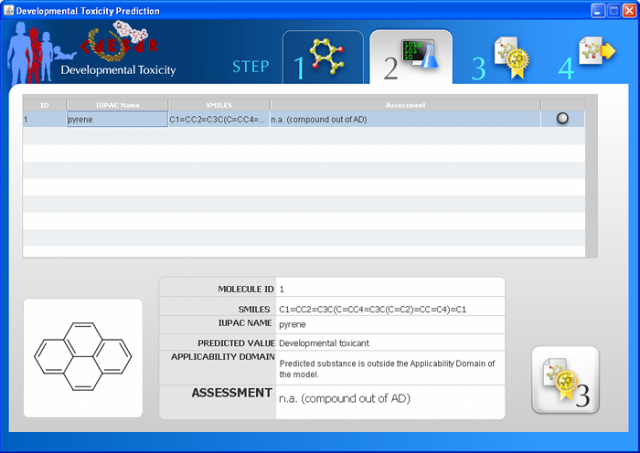
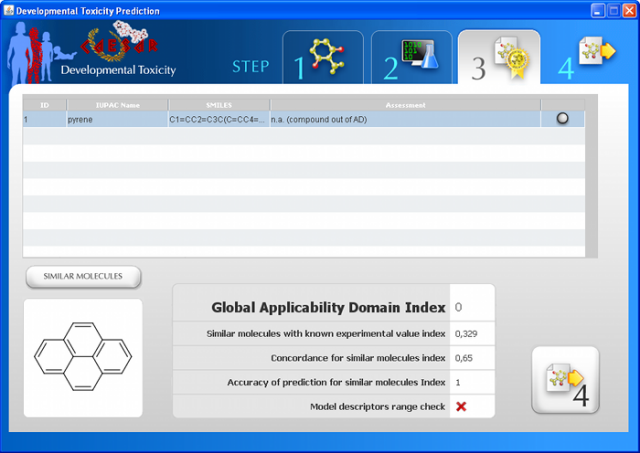


 WARNING: the MUTAGENICITY model could classify a compound as mutagen even if it is formally out of the applicability domain. This behaviour is normal for this model and it is related to the use of structural alerts
WARNING: the MUTAGENICITY model could classify a compound as mutagen even if it is formally out of the applicability domain. This behaviour is normal for this model and it is related to the use of structural alerts 











